More than 40 presentations including data from two pivotal trials selected as late-breaking abstracts for oral presentation in the ESMO Presidential Symposia
Phase III PACIFIC data reinforce potential of IMFINZI®(durvalumab) following US FDA Breakthrough Therapy Designation in locally-advanced (Stage III) unresectable non-small cell lung cancer (NSCLC)
Phase III FLAURA results highlight superiority of TAGRISSO® (osimertinib) over standard of care in previously-untreated patients with advanced EGFR mutation-positive NSCLC
At the European Society of Medical Oncology (ESMO) 2017 Congress in Madrid, Spain, September 8-12, AstraZeneca and MedImmune, its global biologics research and development arm, will report results of more than 40 presentations, including two pivotal clinical trial readouts selected for late-breaking abstract presentation at the ESMO Presidential Symposia on Saturday, September 9, which demonstrate significant improvements over current standard-of-care treatments in lung cancer:
- Results from the investigational, pivotal Phase III PACIFIC trial showing statistically-significant and clinically-meaningful progression-free survival (PFS) benefit with IMFINZI® (durvalumab) in patients with locally-advanced (Stage III), unresectable non-small cell lung cancer (NSCLC) following concurrent chemoradiation therapy, a clinical setting where there are currently no approved treatments.
- Results of the investigational, Phase III FLAURA trial showing statistically-significant and clinically-meaningful PFS benefit with TAGRISSO® (osimertinib) over current standard-of-care erlotinib or gefitinib as 1st-line treatments in previously-untreated patients with locally-advanced or metastatic epidermal growth factor receptor mutation-positive (EGFRm) NSCLC.
Jamie Freedman, Executive Vice President, Head of the Oncology Business Unit at AstraZeneca, said: “The superiority of 1st-line treatment with TAGRISSO in the FLAURA trial and the potentially transformative IMFINZI data from the PACIFIC trial reinforce our significant contribution to advancing medicines for patients across multiple stages of lung cancer. We are also proud to share with the medical community our progress towards addressing the needs of patients with other types of difficult-to-treat tumors, including advanced ovarian, breast, and head and neck cancers.”
Pushing the boundaries of lung cancer research
In addition to the two Presidential Symposia presentations, investigational data being presented at ESMO demonstrate the breadth and depth of AstraZeneca’s commitment to advancing the treatment of lung cancer:
- Overall survival data on osimertinib from the Phase II AURA trials in patients with EGFR T790M mutation-positive advanced or metastatic NSCLC (Abstract #1348P)
- PFS data and central nervous system (CNS) responses to osimertinib in patients with EGFR T790M NSCLC in the Asia Pacific region (AURA17) (Abstracts #1331P, 1353P and 1354P)
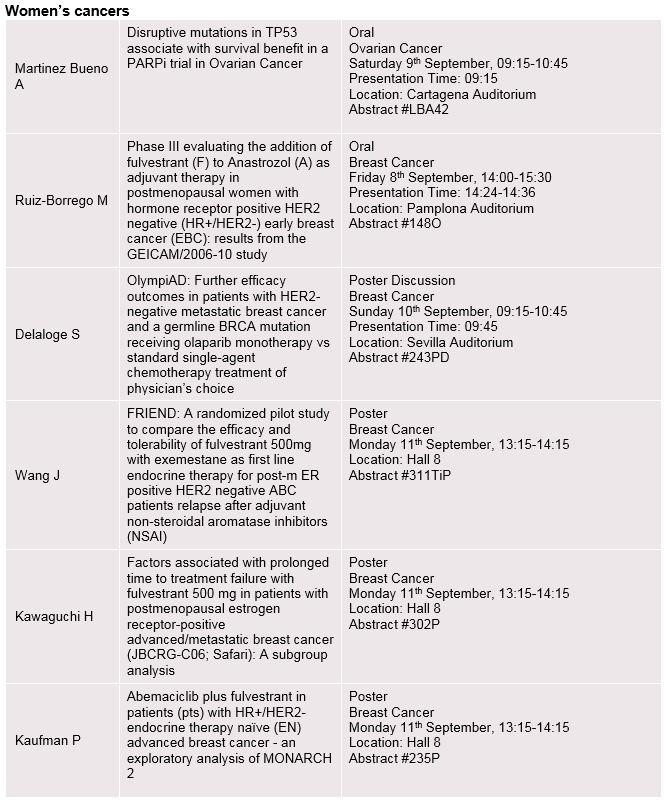
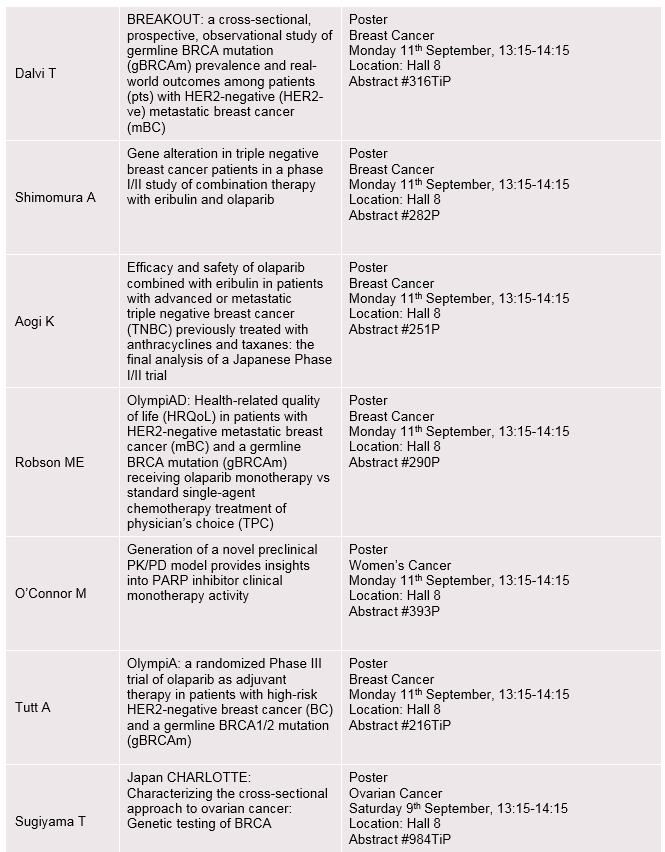
Established expertise in women’s cancers
Emerging investigational data from studies of LYNPARZA® (olaparib), FASLODEX® (fulvestrant), potential new medicines and combinations within women’s cancers will include:
- Latest Phase III OlympiAD efficacy and health-related quality-of-life data for olaparib vs. chemotherapy in patients with HER2-negative metastatic breast cancer and a germline BRCA mutation (Abstract #243PD and 290P)
- Phase III SOLO-2 data for olaparib maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer (Abstract #932PD)
Sustained commitment in head and neck cancer
AstraZeneca is presenting additional evidence of the efficacy and safety of durvalumab and the early potential of its combination strategy in head and neck cancer, including oral presentations on:
- Preliminary investigational data from the Phase II HAWK study of durvalumab in recurrent/metastatic head and neck squamous cell carcinoma (HNSCC) (Abstract #10420)
- Safety, tolerability and anti-tumor activity in the Phase Ib/II SCORES study of durvalumab in combination with STAT3 inhibitor AZD9150 or CXCR2 inhibitor AZD5069, in patients with HNSCC (Abstract #1049PD)
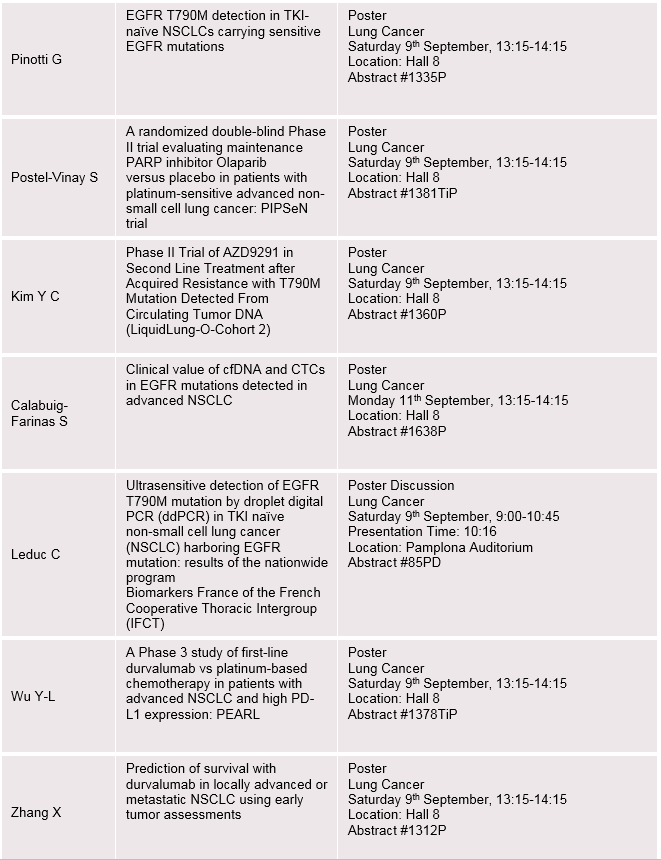
AstraZeneca/MedImmune key presentations at the ESMO 2017 Congress





TAGRISSO® (osimertinib) Important Safety Information
- There are no contraindications for TAGRISSO
- Interstitial Lung Disease (ILD)/Pneumonitis occurred in 3.5% and was fatal in 0.6% of 833 TAGRISSO-treated patients. Withhold TAGRISSO and promptly investigate for ILD in patients who present with worsening of respiratory symptoms indicative of ILD (eg, dyspnea, cough, and fever). Permanently discontinue TAGRISSO if ILD is confirmed
- Heart rate-corrected QT (QTc) interval prolongation occurred in TAGRISSO-treated patients. Of the 833 TAGRISSO-treated patients, 0.7% of patients were found to have a QTc > 500 msec, and 2.9% of patients had an increase from baseline QTc > 60 msec. No QTc-related arrhythmias were reported. Conduct periodic monitoring with ECGs and electrolytes in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities, or those who are taking medications known to prolong the QTc interval. Permanently discontinue TAGRISSO in patients who develop QTc interval prolongation with signs/symptoms of life-threatening arrhythmia
- Cardiomyopathy occurred in 1.9% and was fatal in 0.1% of 833 TAGRISSO-treated patients. Left Ventricular Ejection Fraction (LVEF) decline ≥ 10% and a drop to < 50% occurred in 4% of 655 TAGRISSO-treated patients. Conduct cardiac monitoring, including an assessment of LVEF at baseline and during treatment in patients with cardiac risk factors. Assess LVEF in patients who develop relevant cardiac signs or symptoms during treatment. For symptomatic congestive heart failure or persistent, asymptomatic LV dysfunction that does not resolve within 4 weeks, permanently discontinue TAGRISSO
- Keratitis was reported in 0.7% of 833 TAGRISSO-treated patients in clinical trials. Promptly refer patients with signs and symptoms suggestive of keratitis (such as eye inflammation, lacrimation, light sensitivity, blurred vision, eye pain, and/or red eye) to an ophthalmologist
- Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during TAGRISSO treatment and for 6 weeks after the final dose. Advise males with female partners of reproductive potential to use effective contraception for 4 months after the final dose
- The most common adverse reactions (≥20%) in patients treated with TAGRISSO were diarrhea (41%), rash (34%), dry skin (23%), nail toxicity (22%), and fatigue (22%)
APPROVED USE for TAGRISSO
TAGRISSO is indicated for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation-positive non-small cell lung cancer (NSCLC), as detected by an FDA-approved test, whose disease has progressed on or after EGFR tyrosine kinase inhibitor therapy.
Please see complete Prescribing Information including Patient Information.
IMFINZI® (durvalumab) IMPORTANT SAFETY INFORMATION
There are no contraindications for IMFINZI® (durvalumab).
Monitor patients for clinical signs and symptoms of immune-mediated pneumonitis, hepatitis, colitis or diarrhea, endocrinopathies, nephritis, rash or dermatitis, other immune-mediated adverse reactions, and infection. Please refer to the full Prescribing Information for important dose management information specific to adverse reactions.
Immune-Mediated Pneumonitis
In the combined safety database (n=1414), immune-mediated pneumonitis occurred in 32 patients (2.3%), including 1 fatal case (0.1%) and 6 Grade 3–4 cases (0.4%). In Study 1 (n=182), 1 patient (0.5%) died from immune-mediated pneumonitis. Monitor patients for signs and symptoms of pneumonitis and evaluate with radiographic imaging when suspected. Administer corticosteroids for ≥Grade 2 pneumonitis. Withhold IMFINZI for Grade 2 pneumonitis; permanently discontinue for Grade 3–4 pneumonitis.
Immune-Mediated Hepatitis
In the combined safety database (n=1414), immune-mediated hepatitis occurred in 16 patients (1.1%), including 1 fatal case (<0.1%) and 9 Grade 3 cases (0.6%). Grade 3–4 elevations in ALT occurred in 40/1342 patients (3.0%), AST in 58/1336 patients (4.3%), and total bilirubin in 37/1341 patients (2.8%). In Study 1 (n=182), 1 patient (0.5%) died from immune-mediated hepatitis, and 2 patients (1.1%) experienced immune-mediated hepatitis, including 1 Grade 3 case (0.5%). Monitor patients for abnormal liver tests in each cycle during treatment with IMFINZI. Administer corticosteroids and withhold IMFINZI for Grade 2–3 ALT or AST >3–5X ULN or <8X ULN or total bilirubin >1.5–3X ULN or <5X ULN. Permanently discontinue IMFINZI in patients with Grade 3 ALT or AST >8X ULN or total bilirubin >5X ULN, or in patients with concurrent ALT or AST >3X ULN and total bilirubin >2X ULN with no other cause.
Immune-Mediated Colitis
In the combined safety database (n=1414), immune-mediated colitis or diarrhea occurred in 18 patients (1.3%), including 1 Grade 4 case (<0.1%) and 4 Grade 3 cases (0.3%). In Study 1 (n=182), 23 patients (12.6%) experienced colitis or diarrhea, including 2 Grade 3–4 cases (1.1%). Monitor patients for signs and symptoms of colitis or diarrhea. Administer corticosteroids for ≥Grade 2 colitis or diarrhea. Withhold IMFINZI for Grade 2 colitis or diarrhea; permanently discontinue for Grade 3–4 colitis or diarrhea.
Immune-Mediated Endocrinopathies
- Immune-mediated thyroid disorders, adrenal insufficiency, type 1 diabetes mellitus and hypophysitis/hypopituitarism have occurred with IMFINZI. Monitor patients for clinical signs and symptoms of endocrinopathies. For Grade 2–4 endocrinopathies (except hypothyroidism) withhold dose until clinically stable and offer symptomatic management for hyperthyroidism. For Grade 2–4 hypothyroidism, initiate thyroid hormone replacement as needed
- Thyroid disorders— In the combined safety database (n=1414), immune-mediated hypothyroidism and hyperthyroidism occurred in 136 patients (9.6%) and 81 patients (5.7%), respectively. Thyroiditis occurred in 10 patients (0.7%), including 1 Grade 3 case (<0.1%) in a patient who had a myocardial infarction. In 9 patients with thyroiditis, transient hyperthyroidism preceded hypothyroidism. Treatment with a beta-blocker and/or thioamide was administered for hyperthyroidism in five of these patients. In Study 1 (n=182), Grade 1–2 hypothyroidism or thyroiditis occurred in 10 patients (5.5%). Grade 1–2 hyperthyroidism or thyroiditis leading to hyperthyroidism occurred in 9 patients (4.9%). Monitor patients for abnormal thyroid function tests prior to and periodically during treatment
- Immune-mediated adrenal insufficiency—In the combined safety database (n=1414), immune-mediated adrenal insufficiency occurred in 13 patients (0.9%), including 2 Grade 3 cases (0.1%). In Study 1 (n=182), Grade 1 adrenal insufficiency occurred in 1 patient (0.5%). Administer corticosteroids and hormone replacement as clinically indicated
- Type 1 diabetes mellitus—In the combined safety database (n=1414), new onset type 1 diabetes mellitus without an alternative etiology occurred in 1 patient (<0.1%). For type 1 diabetes mellitus, initiate insulin as indicated and withhold IMFINZI until clinically stable
- Hypophysitis—In the combined safety database (n=1414), hypopituitarism leading to adrenal insufficiency and diabetes insipidus occurred in 1 patient (<0.1%). Administer corticosteroids and hormone replacement as clinically indicated
Other Immune-Mediated Adverse Reactions
- IMFINZI has caused immune-mediated rash. Other immune-related adverse reactions, including aseptic meningitis, hemolytic anemia, immune thrombocytopenic purpura, myocarditis, myositis, nephritis, and ocular inflammatory toxicity including uveitis and keratitis, have occurred in ≤1.0% of patients treated with IMFINZI
- Immune-mediated rash or dermatitis—In the combined safety database (n=1414), immune-mediated rash or dermatitis occurred in 220 patients (15.6%) and 4 patients (0.3%) developed vitiligo. In Study 1 (n=182), 20 patients (11.0%) developed rash, including 1 Grade 3 case (0.5%). Patients should be monitored for signs and symptoms of rash or dermatitis. Administer corticosteroids if indicated. Withhold IMFINZI for Grade 3 rash or dermatitis or Grade 2 rash or dermatitis lasting >1 week. Permanently discontinue IMFINZI in patients with Grade 4 rash or dermatitis
- Immune thrombocytopenic purpura—In the combined safety database (n=1414), 1 fatal case (<0.1%) of immune thrombocytopenic purpura occurred. Monitor patients for signs and symptoms of immune thrombocytopenic purpura
- Nephritis—In the combined safety database (n=1414), immune-mediated nephritis occurred in 3 patients (0.2%), including 2 Grade 3 cases (0.1%). Monitor patients for abnormal renal function tests prior to and during each cycle of IMFINZI. Administer corticosteroids for ≥Grade 2 nephritis (creatinine >1.5X ULN). Withhold IMFINZI for Grade 2 nephritis; permanently discontinue for ≥Grade 3 nephritis (creatinine >3X ULN)
Infection
Severe infections, including sepsis, necrotizing fasciitis, and osteomyelitis, occurred in patients receiving IMFINZI. In the combined safety database (n=1414), infections occurred in 531 patients (37.6%). In Study 1 (n=182), infections occurred in 54 patients (29.7%). 11 patients (6.0%) experienced Grade 3–4 infection and 5 patients (2.7%) were experiencing infection at the time of death. 8 patients (4.4%) experienced urinary tract infection, the most common ≥Grade 3 infection. Monitor patients for signs and symptoms of infection and treat with anti-infectives for suspected or confirmed infections. Withhold IMFINZI for ≥Grade 3 infection.
Infusion-Related Reactions
In the combined safety database (n=1414), severe infusion-related reactions occurred in 26 patients (1.8%). In Study 1 (n=182), infusion-related reactions occurred in 3 patients (1.6%). There were 5 Grade 3 (0.4%) and no Grade 4 or 5 reactions. Patients should be monitored for signs and symptoms of infusion-related reactions. Interrupt or slow the rate of infusion for Grade 1–2 infusion-related reactions and permanently discontinue for Grade 3–4 infusion-related reactions.
Embryo-Fetal Toxicity
Based on its mechanism of action and data from animal studies, IMFINZI can cause fetal harm when administered to a pregnant woman. There are no data on the use of IMFINZI in pregnant women. Advise pregnant women of the potential risk to a fetus and advise women of reproductive potential to use effective contraception during treatment and for at least 3 months after the last dose of IMFINZI.
Nursing Mothers
There is no information regarding the presence of IMFINZI in human milk; however, because of the potential for adverse reactions in breastfed infants from IMFINZI, advise a lactating woman not to breastfeed during treatment and for at least 3 months after the last dose.
Most Common Adverse Reactions
- The most common adverse reactions (≥15%) were fatigue (39%), musculoskeletal pain (24%), constipation (21%), decreased appetite (19%), nausea (16%), peripheral edema (15%), and urinary tract infection (15%). The most common Grade 3 or 4 adverse reactions (≥3%) were fatigue, urinary tract infection, musculoskeletal pain, abdominal pain, dehydration, and general physical health deterioration
- Adverse reactions leading to discontinuation of IMFINZI occurred in 3.3% of patients. Serious adverse reactions occurred in 46% of patients. The most frequent serious adverse reactions (>2%) were acute kidney injury (4.9%), urinary tract infection (4.4%), musculoskeletal pain (4.4%), liver injury (3.3%), general physical health deterioration (3.3%), sepsis, abdominal pain, and pyrexia/tumor associated fever (2.7% each)
The safety and effectiveness of IMFINZI have not been established in pediatric patients.
Approved Uses for IMFINZI
IMFINZI is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who:
- have disease progression during or following platinum-containing chemotherapy
- have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy
This indication is approved under accelerated approval based on tumor response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Please see complete Prescribing Information including Patient Information (Medication Guide).
LYNPARZA® (olaparib) IMPORTANT SAFETY INFORMATION
DOSING AND ADMINISTRATION
To avoid substitution errors and overdose, do not substitute LYNPARZA tablets with LYNPARZA capsules on a milligram-to-milligram basis due to differences in the dosing and bioavailability of each formulation. Recommended tablet dose is 300 mg, taken orally twice daily, with or without food. Continue treatment until disease progression or unacceptable toxicity. For adverse reactions, consider dose interruption or dose reduction.
WARNINGS AND PRECAUTIONS
There are no contraindications for LYNPARZA.
Myelodysplastic Syndrome/Acute Myeloid Leukemia (MDS/AML): Occurred in <1.5% of patients exposed to LYNPARZA monotherapy, and the majority of events had a fatal outcome. The duration of therapy in patients who developed secondary MDS/AML varied from <6 months to >2 years. All of these patients had previous chemotherapy with platinum agents and/or other DNA-damaging agents, including radiotherapy, and some of these patients also had a history of previous cancer or bone marrow dysplasia.
Do not start LYNPARZA until patients have recovered from hematological toxicity caused by previous chemotherapy (≤Grade 1). Monitor complete blood counts for cytopenia at baseline and monthly thereafter for clinically significant changes during treatment. For prolonged hematological toxicities, interrupt LYNPARZA and monitor blood counts weekly until recovery. If the levels have not recovered to Grade 1 or less after 4 weeks, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. Discontinue LYNPARZA if MDS/AML is confirmed.
Pneumonitis: Occurred in <1% of patients exposed to LYNPARZA, and some cases were fatal. If patients present with new or worsening respiratory symptoms such as dyspnea, cough, and fever, or a radiological abnormality occurs, interrupt treatment with LYNPARZA and initiate prompt investigation. Discontinue LYNPARZA if pneumonitis is confirmed and treat patient appropriately.
Embryo-Fetal Toxicity: Based on its mechanism of action and findings in animals, LYNPARZA can cause fetal harm. A pregnancy test is recommended for females of reproductive potential prior to initiating treatment. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for 6 months after receiving the final dose.
ADVERSE REACTIONS—Maintenance Setting
Most common adverse reactions (Grades 1-4) in ≥20% of patients in clinical trials of LYNPARZA in the maintenance setting for SOLO-2: nausea (76%), fatigue (including asthenia) (66%), anemia (44%), vomiting (37%), nasopharyngitis/upper respiratory tract infection (URI)/influenza (36%), diarrhea (33%), arthralgia/myalgia (30%), dysgeusia (27%), headache (26%), decreased appetite (22%), and stomatitis (20%).
Study 19: nausea (71%), fatigue (including asthenia) (63%), vomiting (35%), diarrhea (28%), anemia (23%), respiratory tract infection (22%), constipation (22%), headache (21%), and decreased appetite (21%).
Most common laboratory abnormalities (Grades 1-4) in ≥25% of patients in clinical trials of LYNPARZA in the maintenance setting (SOLO-2/Study 19) were: increase in mean corpuscular volume (89%/82%), decrease in hemoglobin (83%/82%), decrease in leukocytes (69%/58%), decrease in lymphocytes (67%/52%), decrease in absolute neutrophil count (51%/47%), increase in serum creatinine (44%/45%), and decrease in platelets (42%/36%).
ADVERSE REACTIONS—Advanced gBRCAm ovarian cancer
Most common adverse reactions (Grades 1-4) in ≥20% of patients in clinical trials of LYNPARZA for advanced gBRCAm ovarian cancer after 3 or more lines of chemotherapy (pooled from 6 studies) were: fatigue (including asthenia) (66%), nausea (64%), vomiting (43%), anemia (34%), diarrhea (31%), nasopharyngitis/upper respiratory tract infection (URI) (26%), dyspepsia (25%), myalgia (22%), decreased appetite (22%), and arthralgia/musculoskeletal pain (21%).
Most common laboratory abnormalities (Grades 1-4) in ≥25% of patients in clinical trials of LYNPARZA for advanced gBRCAm ovarian cancer after 3 or more lines of chemotherapy (pooled from 6 studies) were: decrease in hemoglobin (90%), increase in mean corpuscular volume (57%), decrease in lymphocytes (56%), increase in serum creatinine (30%), decrease in platelets (30%), and decrease in absolute neutrophil count (25%).
DRUG INTERACTIONS
Anticancer Agents: Clinical studies of LYNPARZA in combination with other myelosuppressive anticancer agents, including DNA-damaging agents, indicate a potentiation and prolongation of myelosuppressive toxicity.
CYP3A Inhibitors: Avoid concomitant use of strong or moderate CYP3A inhibitors. If a strong or moderate CYP3A inhibitor must be co-administered, reduce the dose of LYNPARZA. Advise patients to avoid grapefruit, grapefruit juice, Seville oranges, and Seville orange juice during LYNPARZA treatment.
CYP3A Inducers: Avoid concomitant use of strong or moderate CYP3A inducers when using LYNPARZA. If a moderate inducer cannot be avoided, be aware of a potential for decreased efficacy of LYNPARZA.
USE IN SPECIFIC POPULATIONS
Pediatric Use: The safety and efficacy of LYNPARZA have not been established in pediatric patients.
Lactation: No data are available regarding the presence of olaparib in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed infant, advise a lactating woman not to breastfeed during treatment with LYNPARZA and for 1 month after receiving the final dose.
Hepatic Impairment: No adjustment to the starting dose is required in patients with mild hepatic impairment (Child-Pugh classification A). There are no data in patients with moderate or severe hepatic impairment.
Renal Impairment: No adjustment to the starting dose is necessary in patients with mild renal impairment (CLcr 51-80 mL/min). In patients with moderate renal impairment (CLcr 31-50 mL/min), reduce the dose to 200 mg twice daily. There are no data in patients with severe renal impairment or end-stage renal disease (CLcr ≤30 mL/min).
APPROVED USES for LYNPARZA
LYNPARZA is a poly (ADP-ribose) polymerase (PARP) inhibitor indicated:
- For the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete or partial response to platinum-based chemotherapy
- For the treatment of adult patients with deleterious or suspected deleterious germline BRCA-mutated advanced ovarian cancer who have been treated with 3 or more prior lines of chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA
Please see complete Prescribing Information, including Patient Information (Medication Guide).
FASLODEX® (fulvestrant) IMPORTANT SAFETY INFORMATION
Contraindications
- FASLODEX is contraindicated in patients with known hypersensitivity to the drug or to any of its components. Hypersensitivity reactions, including urticaria and angioedema, have been reported in association with FASLODEX
Risk of Bleeding
- Because FASLODEX is administered intramuscularly, it should be used with caution in patients with bleeding diatheses, thrombocytopenia, or anticoagulant use
Hepatic Impairment
- FASLODEX is metabolized primarily in the liver. A 250-mg dose is recommended in patients with moderate hepatic impairment (Child-Pugh class B). FASLODEX has not been evaluated in patients with severe hepatic impairment (Child-Pugh class C)
Injection Site Reaction
- Use caution while administering FASLODEX at the dorsogluteal injection site due to the proximity of the underlying sciatic nerve. Injection site related events including sciatica, neuralgia, neuropathic pain, and peripheral neuropathy have been reported with FASLODEX injection
Embryo-Fetal Toxicity and Lactation
- Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during FASLODEX treatment and for 1 year after the final dose. Advise lactating women not to breastfeed during treatment with FASLODEX and for 1 year after the final dose because of the potential risk to the infant
Immunoassay Measurement of Serum Estradiol
- Due to structural similarity of fulvestrant and estradiol, FASLODEX can interfere with estradiol measurement by immunoassay, resulting in falsely elevated estradiol levels
Adverse Reactions
Monotherapy
- The most common adverse reactions occurring in ≥5% of patients receiving FASLODEX 500 mg were: injection site pain, nausea, bone pain, arthralgia, headache, back pain, fatigue, pain in extremity, hot flash, myalgia, vomiting, anorexia, diarrhea, asthenia, musculoskeletal pain, cough, dyspnea, and constipation
- Increased hepatic enzymes (ALT, AST, ALP) occurred in >15% of FASLODEX patients and were not dose-dependent
Combination Therapy
- The most frequently reported serious adverse reactions in patients receiving FASLODEX plus palbociclib were infections (3%), pyrexia (1%), neutropenia (1%), and pulmonary embolism (1%)
- The most common adverse reactions (≥10%) of any grade reported in patients receiving FASLODEX 500 mg plus palbociclib 125 mg/day were: neutropenia, leukopenia, infections, fatigue, nausea, anemia, stomatitis, headache, diarrhea, thrombocytopenia, constipation, vomiting, alopecia, rash, decreased appetite, and pyrexia
APPROVED USES for FASLODEX
Monotherapy
FASLODEX is an estrogen receptor antagonist indicated for the:
- Treatment of hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced breast cancer in postmenopausal women not previously treated with endocrine therapy
- Treatment of HR-positive advanced breast cancer in postmenopausal women with disease progression following endocrine therapy
Combination Therapy
- FASLODEX in combination with palbociclib is indicated for the treatment of HR-positive, HER2-negative advanced or metastatic breast cancer in women with disease progression after endocrine therapy
Please see full Prescribing Information with Patient Information.
NOTES TO EDITORS
About AstraZeneca in Oncology
AstraZeneca has a deep-rooted heritage in Oncology and offers a quickly growing portfolio of new medicines that has the potential to transform patients’ lives and the Company’s future. With at least six new medicines to be launched between 2014 and 2020 and a broad pipeline of small molecules and biologics in development, we are committed to advance New Oncology as one of AstraZeneca’s five Growth Platforms focused on lung, ovarian, breast and blood cancers. In addition to our core capabilities, we actively pursue innovative partnerships and investments that accelerate the delivery of our strategy as illustrated by our investment in Acerta Pharma in hematology.
By harnessing the power of four scientific platforms – Immuno-Oncology, Tumor Drivers and Resistance, DNA Damage Response and Antibody Drug Conjugates – and by championing the development of personalized combinations, AstraZeneca has the vision to redefine cancer treatment and one day eliminate cancer as a cause of death.
About MedImmune
MedImmune is the global biologics research and development arm of AstraZeneca, a global, innovation-driven biopharmaceutical business that focuses on the discovery, development and commercialization of small molecule and biologic prescription medicines. MedImmune is pioneering innovative research and exploring novel pathways across Oncology; Respiratory, Cardiovascular & Metabolic Diseases; and Infection and Vaccines. The MedImmune headquarters is located in Gaithersburg, MD, one of AstraZeneca’s three global R&D centers, with additional sites in Cambridge, UK, and Mountain View, CA. For more information, please visit www.medimmune.com.
About AstraZeneca
AstraZeneca is a global, science-led biopharmaceutical company that focuses on the discovery, development and commercialization of prescription medicines, primarily for the treatment of diseases in three main therapy areas – Oncology, Cardiovascular & Metabolic Diseases and Respiratory. The Company also is selectively active in the areas of autoimmunity, neuroscience and infection. AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. For more information, please visit www.astrazeneca-us.com and follow us on Twitter @AstraZenecaUS.
CONTACTS
Media Inquiries
Michele Meixell +1 302 885 2677
Alexandra Engel +1 302 885 2677
US-14081 Last Updated 8/17